기대 밑도는 '레켐비' 투약 속 바이오젠 매출 제자리
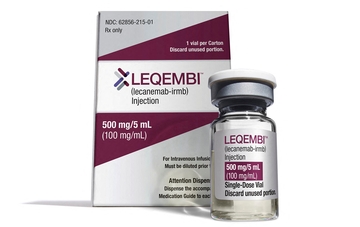
메디칼타임즈=문성호 기자알츠하이머 치료 신약으로 기대 받던 레켐비(레카네맙)의 실적이 기대에 못 미치는 모양새다. 실제 투여환자가 기대 이하이기 때문인데 회사 측은 낙관적인 반응을 내놨다.바이오젠 알츠하이머 치료제 레켐비 제품사진.15일 제약업계에 따르면, 최근 바이오젠은 실적발표를 통해 지난해 4분기 매출이 전년 동기 대비 6.2% 감소한 23억 9000만달러(약 3조 1900억원)를 기록했다고 발표했다. 연간 총 매출은 3% 감소한 98억 달러(약 13조원)로 집계됐다.이 가운데 관심을 끄는 부분은 알츠하이머 치료 신약으로 불리는 '레켐비'의 성과다. 바이오젠과 에자이가 공동 개발한 레켐비는 알츠하이머병 치료를 위한 아밀로이드 베타 항체 기반 치료제다. 알츠하이머병 환자의 뇌속에서는 신경독성을 가진 아밀로이드 베타(Aβ) 축적이 일어나는데 레켐비는 이런 응집을 막는 방식으로 작용한다. 지난해 7월 미국식품의약국(FDA)이 레켐비를 정식 승인하면서 사실상 첫 치매 신약이 탄생했다는 평가가 나오기도 했다.이에 따라 바이오젠은 최근까지 레켐비를 처방받았거나 처방 받을 환자가 3800명으로 추정된다고 설명했다. 올 3월까지 미국에서 레켐비 처방 환자 수 목표를 1만명으로 잡았던 것을 고려하면 기대보다 투여 환자가 적다는 평가가 나올 수 있는 부분이다.임상현장에서는 이 같은 레켐비의 투여에 있어 '가격' 장애물이 발생했다는 평가를 하고 있다.A대학병원 신경과 교수는 "아두카누맙이 처음 등장했을 때 연간 약제비가 약 6600만원으로 책정됐고 이어 반값으로 낮췄지만 상업적 성공은 거두지 못했다"며 "레카네맙의 연간 약제비는 연간 약 3300만원 수준이 될 것으로 전망되는데 이마저도 효과에 비하면 과도한 측면이 없잖아 있다"고 지적했다.그는 "연간 수 천만원을 지불할 능력이 있는 환자군은 많지 않기 때문에 보험 등재 시 약가 조정이 처방 활성화의 관건"이라며 "합리적인 가격으로 국내에 들어온다면 처방 수요는 충분하다고 본다"고 전망했다. 하지만 바이오젠 측은 향후 레켐비의 활용이 늘어날 것이란 전망을 내놨다.바이오젠 비바커(Viehbacher) CEO는 "치료제 수요가 있다는 것은 분명하다"며 "레켐비 판매는 점진적으로 올라갈 것이며, 이는 장기적으로 봤을 때 매우 중요하다"고 평가했다.이어 "2023년은 치료제 4종을 승인받았고, 비용 구조를 재조정했다"며 "최근 신제품을 출시하고 있는 상황으로 효율적인 운영으로 성과를 창출하겠다"고 말했다.